ICM Web: The Interactive
Chromatin Modeling Webserver
Richard C. Stolz and Thomas
C. Bishop
Louisiana Tech University
Departments of Chemistry and Physics
College of Engineering & Science
Ruston, LA 71272, U.S.A.
Tel.: (318)257-5209, Fax: (318)257-4000
bishop@latech.edu
Index:
Link to Nucleic Acids Res. 2010 38 W254: The
Interactive Chromatin Modeling Webserver
Overview
Instructions
Sequence
Selection
Energy
Selection
Sample Output
Algorithm
Related Links
Overview
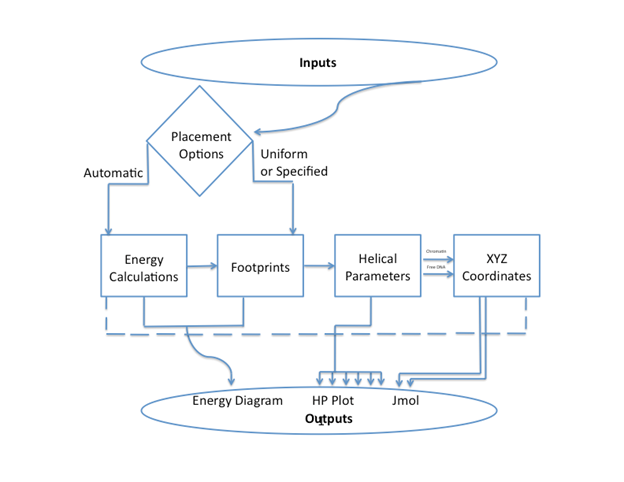
General Usage: ICM-Web requires the user to input a sequence of DNA, select options for thermal fluctuation and
placement then click "Go". ICM
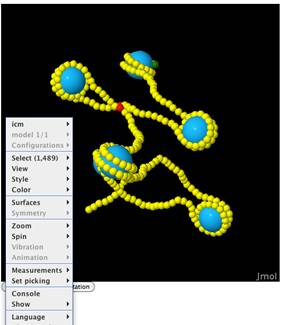
folds the sequence into a coarse-grain model of free DNA and a nucleosome array. The results page includes an energy level
diagram, helical parameter plots, 3D
molecular graphics displayed in Jmol, and options for saving data.
The following pages describe:
Sequence
Input Options
Energy
Selection
Back to
Top
Instructions
Sequence Input Options:
There are three options in this panel. Select “Default”, which will use the default
sequence, “Type Sequence”, which will allow a sequence to either be typed, or
cut and pasted into a text box, or “Upload Sequence” which accepts FASTA
formatted sequence files (without the header) for upload.
|

|
|
Default Sequence:
Using this option the MMTV sequence is used
in calculations. Continue onto Energy
Selection from here.
|
|
|
|
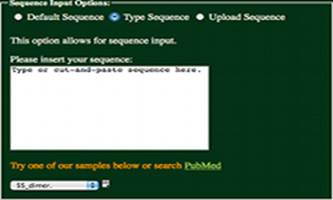
Type Sequence:
Having selected the “Type Sequence” radio
button, a sequence may be entered into the text box as shown to the left.
|
|
|
|
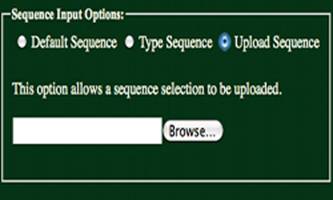
Upload Sequence:
Having selected the “Upload Sequence” radio
button, a sequence may be uploaded using the form shown to the left. Uploaded files maybe in FASTA format.
Headers from FASTA formatted files must be removed. Only ‘A’, ‘T’, ‘G’, &’C’ characters
will be accepted (either uppercase or lowercase). All other characters will return an error.
|
Back to
Top
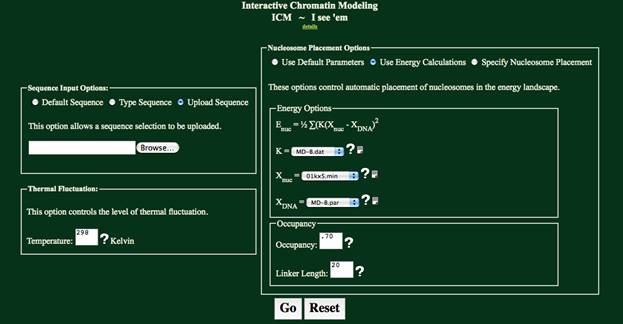
Nucleosome Placement Options:
There are three choices in placing nucelosomes in ICM. Select “Use Default Parameters”, which will
use the default energy profile, “Use Energy Calculations”, which will allow an
energy profile to be specified, or “Specify Nucleosome Placement” which accepts
Nucleosome Start Sites as the placement parameter for the nucleosome.
|

|
|
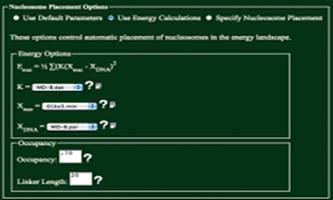
Use Default Parameters
Using
this option will use Xnuc=MD-B.par, Xnuc=01kxf.min, K = MD-B.dat, an occupancy of .7, and a
linker of 20. This option was
developed as to help those not familiar with the parameters used in
calculations to get a general feel for the folding of DNA.
|
|
|
|
Use Energy Calculations
Using
this option will allows for Xnuc, Xnuc, K, occupancy,
and linker values to be specified.
These are described in further detail below under the “Energy Options”
heading.
|
|

|
|
Specify
Nucleosome Placement
Using this option will allow nucleosome start
sites to be specified. K, XDNA, and
Xnuc values are all defined as in the “Use Energy Calculations”
option. However, these values are used
only in making the energy graph, not used in placing the nucleosomes. Only the specified start sites are used in
placing the nucleosomes.
|
Back to
Top
Energy Options:
The “Energy Options” box is provided for the “Use Energy
Calculations” option and the “Specify Nucleosome Start Sites” option. The “Use Energy Calculations” option uses
these parameters in determining the nucleosome positioning, while the “Specify
Nucleosome Start Sites” uses these parameters only in creating an energy plot.

K is the measure of the
stiffness of DNA. It accounts for six
degrees of freedom: Shift, Slide, Rise, Tilt, Roll, and Twist.
Xnuc is the shape
of the nucleosome. This button lets you
select from any of the available x-ray structures. All parameters extracted using 3DNA.
Xdna is the shape
of the DNA when free in solution. This
button lets you define the shape of free DNA to agree with x-ray or MD data.
Thermal
Fluctuation
This determines the thermal fluctuations in the helical parameters
for the regions of free DNA. The
fluctuations yield a Gaussian distribution as determined by the chosen
stiffness parameters and temperature.

Back to
Top
Occupancy
& Linker Length
The occupancy determines how many nucleosomes are placed.
n > 1 : integer number of
nucleosomes
n < 1 : percentage of maximum possible number of
nucleosomes
n = 0 : constant spacing of "linker length" between
nucleosomes
n < 0 : constant spacing with all locations shifted by n
The linker length is the minimum nucleosome- nucleosome distance in basepair.

Back to
Top
Sample Output
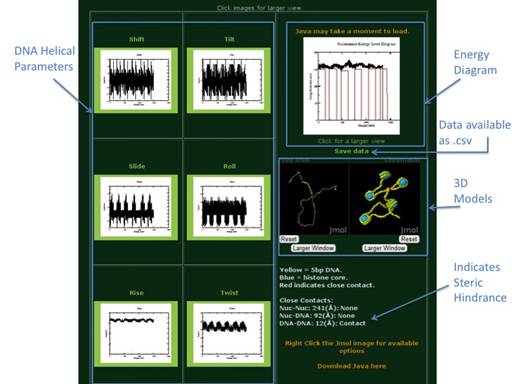 Using the default sequence and
default energy parameters ICM-Web will give the above output. Each output page includes the six DNA Helical
Paramerter (Shift, Slide, Rise, Tilt, Roll, Twist) Plots, an Energy Diagram,
downloadable data, two 3D models (one of free DNA and one of
chromatin), and an indication of wether steric hinderance may be a problem in
the model or not. For more details on
the model’s output please reference the Nucleic Acids Research article
Using the default sequence and
default energy parameters ICM-Web will give the above output. Each output page includes the six DNA Helical
Paramerter (Shift, Slide, Rise, Tilt, Roll, Twist) Plots, an Energy Diagram,
downloadable data, two 3D models (one of free DNA and one of
chromatin), and an indication of wether steric hinderance may be a problem in
the model or not. For more details on
the model’s output please reference the Nucleic Acids Research article
referenced at the
beginning of this
document.